At the point of injury, the concussion epidemic begins with the neurometabolic cascade usually after whiplash, neck injury. We address this root cause of the short and long term concussion symptoms to ensure patients get better faster and healthier. Dr. Ruben St. Laurent is a concussion doctor who addresses mild traumatic brain injuries (mTBI) with a compassionate heart and medical research following the NEI protocols.
The Neurometabolic Cascade of Concussion
Journal of Athletic Training 2001 Jul-Sep; 36(3): 228–235. PMCID: PMC155411
Christopher C. Giza and David A. Hovda
Author information Copyright and License information
This article has been cited by other articles in PMC.
Abstract
Objective:
To review the underlying pathophysiologic processes of concussive brain injury and relate these neurometabolic changes to clinical sports-related issues such as injury to the developing brain, overuse injury, and repeated concussion.
Data Sources:
Over 100 articles from both basic science and clinical medical literature selected for relevance to concussive brain injury, postinjury pathophysiology, and recovery of function.
Data Synthesis:
The primary elements of the pathophysiologic cascade following concussive brain injury include abrupt neuronal depolarization, release of excitatory neurotransmitters, ionic shifts, changes in glucose metabolism, altered cerebral blood flow, and impaired axonal function. These alterations can be correlated with periods of postconcussion vulnerability and with neurobehavioral abnormalities. While the time course of these changes is well understood in experimental animal models, it is only beginning to be characterized following human concussion.
Conclusions/Recommendations:
Following concussion, cerebral pathophysiology can be adversely affected for days in animals and weeks in humans. Significant changes in cerebral glucose metabolism can exist even in head-injured patients with normal Glasgow Coma Scores, underscoring the need for in-depth clinical assessment in an effort to uncover neurocognitive correlates of altered cerebral physiology. Improved guidelines for clinical management of concussion may be formulated as the functional significance and duration of these postinjury neurometabolic derangements are better delineated.
Concussion is defined as any transient neurologic dysfunction resulting from a biomechanical force. Loss of consciousness is a clinical hallmark of concussion but is not required to make the diagnosis. Other symptoms include confusion, disorientation, unsteadiness, dizziness, headache, and visual disturbances. These postconcussive deficits occur with minimal detectable anatomic pathology and often resolve completely over time, suggesting that they are based on temporary neuronal dysfunction rather than cell death. Neuronal dysfunction can occur due to ionic shifts, altered metabolism, impaired connectivity, or changes in neurotransmission. Thus, a complete understanding of the phenomenon of concussion requires knowledge of the underlying pathophysiology of this injury. In this article, we will review the neurometabolic events following experimental concussive brain injury and then apply this knowledge to specific scenarios pertinent to sport-related concussion.
POSTCONCUSSIVE PATHOPHYSIOLOGY
An Overview of Concussion Pathophysiology
Immediately after biomechanical injury to the brain, abrupt, indiscriminant release of neurotransmitters and unchecked ionic fluxes occur. The binding of excitatory transmitters, such as glutamate, to the N-methyl-D-aspartate (NMDA) receptor leads to further neuronal depolarization with efflux of potassium and influx of calcium. These ionic shifts lead to acute and subacute changes in cellular physiology.
Acutely, in an effort to restore the neuronal membrane potential, the sodium-potassium (Na+-K+) pump works overtime. The Na+-K+ pump requires increasing amounts of adenosine triphosphate (ATP), triggering a dramatic jump in glucose metabolism. This “hypermetabolism” occurs in the setting of diminished cerebral blood flow, and the disparity between glucose supply and demand triggers a cellular energy crisis. The resulting energy crisis is a likely mechanism for postconcussive vulnerability, making the brain less able to respond adequately to a second injury and potentially leading to longer-lasting deficits.
Following the initial period of accelerated glucose utilization, the concussed brain goes into a period of depressed metabolism. Persistent increases in calcium may impair mitochondrial oxidative metabolism and worsen the energy crisis. Unchecked calcium accumulation can also directly activate pathways leading to cell death. Intra-axonal calcium flux has been shown to disrupt neurofilaments and microtubules, impairing posttraumatic neural connectivity.
This overview represents a simplified framework of the neurometabolic cascade (Figure 1). Other important components of posttraumatic cerebral pathophysiology include, but are not limited to, generation of lactic acid, decreased intracellular magnesium, free radical production, inflammatory responses, and altered neurotransmission. We will now discuss some of the pertinent details of postconcussive pathophysiology in both experimental animal models and in humans.
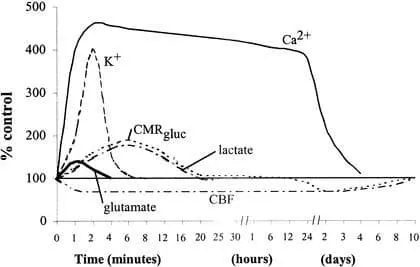
Figure 1: Neurometabolic cascade following experimental concussion. K+, potassium; Ca2+, calcium; CMRgluc, oxidative glucose metabolism; CBF, cerebral blood flow. (Reprinted with permission. Giza CC, Hovda DA. Ionic and metabolic consequences of concussion. In: Cantu RC, Cantu RI. Neurologic Athletic and Spine Injuries. St Louis, MO: WB Saunders Co; 2000:80–100.).
Acute Metabolic and Ionic Changes
Immediately after biomechanical injury to the brain, there is disruption of neuronal membranes, axonal stretching, and opening of voltage-dependent K+ channels, which leads to a marked increase in extracellular K+.1–4 In addition, nonspecific depolarization leads to an early, indiscriminate release of the excitatory amino acid (EAA) glutamate, which exacerbates the K+ flux by activating kainate, NMDA, and D-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA) receptors (Figure 2, events 1 and 2). In rats, treatment with the EAA inhibitor kynurenic acid greatly diminishes the posttraumatic K+ efflux.3
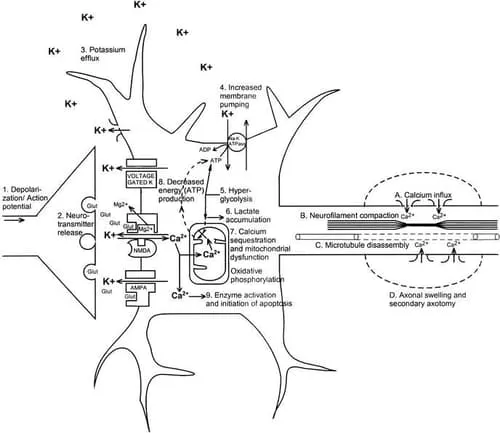
Normally, excessive extracellular K+ is taken up by surrounding glial cells.5–7 By this mechanism, the brain can maintain physiologic K+ levels after mild perturbations; however, larger insults, such as brain trauma or ischemia, overcome this compensation.8–11 As extracellular K+ increases, neuronal depolarization is triggered, leading to further release of EAAs, opening of EAA receptor channels (NMDA, AMPA, kainate), and still greater K+ flux (Figure 2, event 3). This massive excitation is then followed by a wave of relative neuronal suppression that has been termed spreading depression.12–16 One important distinction between classic spreading depression and postconcussive K+ fluxes is that after trauma, diffuse areas of the brain are affected simultaneously. Early loss of consciousness, amnesia, or other cognitive dysfunction may be manifestations of a posttraumatic spreading depression–like state.
In an effort to restore ionic homeostasis, energy-requiring membrane pumps are activated17–19 and trigger an increase in glucose use (Figure 2, events 4 and 5).20–22 This increase in glucose use occurs almost immediately after fluid percussion injury in rats and persists for up to 30 minutes in the ipsilateral cortex and hippocampus.22 After more severe injury such as cortical contusion, increased glucose metabolism may last 4 hours in areas distant from the contusion core.23 Because cerebral oxidative metabolism typically runs near its maximum, an abrupt increase in energy requirements is best met by an increase in glycolysis.24,25
Accelerated glycolysis leads to increased lactate production and is seen after both ischemic26–28 and concussive29–33 brain injury. In addition to hyperglycolysis, oxidative metabolism is also impaired after brain trauma.34–36 This impairment of mitochondrial function can lead to reduced ATP production, which provides a second stimulus for increased glycolysis. Thus, lactate production by glycolysis increases concurrent with a decrease in lactate metabolism, resulting in lactate accumulation (Figure 2, event 6). Elevated lactate levels can result in neuronal dysfunction by inducing acidosis, membrane damage, altered blood brain barrier permeability, and cerebral edema.37–41 Increased levels of lactate after traumatic brain injury (TBI) may leave neurons more vulnerable to a secondary ischemic injury,42 but whether this is the case in repeated traumatic injury is not known. An intriguing hypothesis suggests that glial lactate production increases posttraumatically and that this excess lactate is actually transported into neurons for use as an alternate fuel.43
Cerebral Blood Flow–Glucose Metabolism Uncoupling
Under normal conditions, cerebral blood flow (CBF) is tightly coupled to neuronal activity and cerebral glucose metabolism. After experimental fluid percussion injury, however, CBF may be reduced to 50% of normal.44–47 This posttraumatic decrease in CBF does not approach the 85% reduction seen in frank ischemia48; nonetheless, in a setting of increased glucose use (hyperglycolysis), this mismatch in supply and demand results in a potentially damaging energy crisis.
Calcium Influx, Mitochondrial Dysfunction, and Delayed Glucose Hypometabolism
Calcium accumulation is seen within hours of experimental concussion and may persist for 2 to 4 days.49–52 The posttraumatic depolarization and K+ efflux triggers the release of EAAs that, in turn, activates NMDA receptors.3 Activated NMDA receptors form a pore through which calcium (Ca2+) can enter the cell. A potent N-type calcium channel blocker, SNX-111, significantly reduces postconcussive Ca2+ accumulation,53 presumably by reducing release of glutamate.54 Results of treatment with NMDA receptor antagonists have been mixed, however. No reduction in Ca2+ accumulation was seen after weight-drop injury and pre-treatment with the NMDA receptor blocker MK-801,55 but treatment with HU-211, a synthetic cannabinoid with a pharmacologic profile characteristic of an NMDA receptor antagonist, was associated with a reduction in post-TBI Ca2+ accumulation.56
Excess intracellular Ca2+ may also be sequestered in mitochondria,34,36 resulting in impaired oxidative metabolism and, ultimately, energy failure (Figure 2, events 7 and 8). Cytochrome oxidase histochemistry, which is a measure of oxidative metabolism, shows a biphasic reduction after experimental concussion. In the ipsilateral cortex, a relative reduction on day 1 recovers by day 2, only to be reinstated on day 3, to bottom out on day 5, and to recover by 10 days postinjury. Smaller but more lasting changes are seen in the ipsilateral hippocampus, with decreases still evident at 10 days.57
After the initial period of hyperglycolysis, cerebral glucose use is diminished by 24 hours postinjury and remains low for 5 to 10 days in experimental animals.22 Positron emission tomography (PET) in humans shows similar decreases in global cerebral glucose metabolism that may last 2–4 weeks post-TBI.58 In addition, this study found that post-injury hypometabolism did not correlate closely with level of consciousness as measured by Glasgow Coma Score (GCS). Depressed cerebral glucose metabolism was seen in comatose patients as well as in walking, talking patients, suggesting that significant neurometabolic abnormalities may occur in the absence of overt clinical symptoms.58 What is not yet clear is whether this hypometabolism represents a period when the brain is relatively protected from secondary injury or if the brain is more vulnerable because it is unable to respond adequately to further energy demands. It is also unknown whether this depressed glucose metabolism is responsible for more subtle neurocognitive deficits seen after TBI.
Reductions in Magnesium
Intracellular magnesium levels are also immediately reduced after TBI and remain low for up to 4 days.59–62 This reduction in magnesium has been correlated with postinjury neurologic deficits, and pretreatment to restore magnesium levels results in improved motor performance in experimental animals.63 Decreased magnesium levels may lead to neuronal dysfunction via multiple mechanisms. Both glycolytic and oxidative generation of ATP are impaired when magnesium levels are low. Magnesium is necessary for maintaining the cellular membrane potential and initiating protein synthesis. Finally, low levels of magnesium may effectively unblock the NMDA receptor channel more easily, leading to greater influx of Ca2+ and its potentially deleterious intracellular consequences.
Diffuse Axonal Injury
Mechanical stretching of axons may result in membrane disruption and even depolarization.4 Increased axolemmal permeability persists for up to 6 hours postinjury64,65 and can lead to influx of Ca2+ (Figure 2, event 9A) and mitochondrial swelling.66,67 Neurofilament compaction occurs from 5 minutes to 6 hours postinjury (Figure 2, event 9B), either by phosphorylation, which alters neurofilament stability,68–70 or by calpain-mediated proteolysis of sidearms, which can lead to neurofilament collapse.71 Increased axonal Ca2+ levels have been shown to lead to microtubule breakdown from 6 to 24 hours after the initial injury (Figure 2, event 9C).72,73
Intra-axonal cytoskeletal abnormalities lead to accumulation of organelles at the site of axonal damage due to continued axonal transport along intact segments. These focal axonal swellings eventually develop constrictions that then lead to secondary axotomy (Figure 2, event 9D) and formation of axonal bulbs.73,74 Signs of secondary axonal disconnection may be seen as soon as 4 hours postinjury but have been reported to persist over days and even weeks in brain-injured humans.75
Delayed Cell Death and Persistent Calcium Accumulation
Post-TBI increases in Ca2+ do not inevitably lead to cell death. As mentioned previously, elevated intracellular Ca2+ may certainly lead to impaired mitochondrial metabolism, but neurons may still survive. In fact, after moderate experimental concussion, Ca2+ accumulation peaks in 2 days and resolves without obvious morphologic damage by 4 days.50 Animals experiencing more severe injury and demonstrating anatomic damage show persistent Ca2+ elevations at the injury site. In adult animals, a delayed rise (14 days postinjury) of Ca2+ is seen in distant structures (thalamus), which corresponds to neuronal death.52
Intracellular Ca2+ may trigger cell death by a variety of mechanisms (Figure 2, event 9), including overactivation of phospholipases,76 plasmalogenase, calpains,77,78 protein kinases,79 nitric oxide synthase, and endonucleases. These alterations may then lead to free radical overproduction,80 cytoskeletal reorganization,81 and activation of apoptotic genetic signals.82
Neurotransmitter Alterations
Long-term deficits in memory and cognition in a setting of minimal anatomic change are often seen after concussion. These may result from dysfunctional excitatory neurotransmission. Postconcussive alterations have been reported in glutamatergic (NMDA),83–85 adrenergic,86,87 and cholinergic88 systems. Long-term potentiation, an NMDA-dependent measure of plasticity, may be persistently impaired in the hippocampus after concussive brain injury.89–91 Concussive brain injury also leads to early changes in choline acetyltransferase activity88 and later loss of forebrain cholinergic neurons.92 Impaired cholinergic neurotransmission leads to learning and spatial memory deficits in animals.93,94
Inhibitory neurotransmission is also altered after TBI. A loss of γ-aminobutyric acid-producing (GABAergic) hilar neurons can compromise normal inhibition of hippocampal dentate granule cells.95 This loss of inhibitory neurons may predispose the traumatized brain to subsequent development of seizures.96
CONCERNS RELEVANT TO ATHLETIC CONCUSSION
Concussion in the Developing Brain
With increasing numbers of children and young adults participating in organized sports and sustaining head injuries, understanding the effects of TBI on the immature brain becomes more and more important. Clinical dogma has held that the younger the brain, the more resilient it is after injury. Recent studies of moderate fluid percussion in juvenile rats would seem to support this contention, demonstrating no obvious neurologic or pathologic deficits in these young animals.97,98 Using a closed head injury weight-drop model, significant deficits are seen only at injury severities that result in very high mortality (75%).99
On the other hand, there is also evidence to support the idea of specific developmental periods when the young brain may be more vulnerable to injury. TBI in children results in higher mortality than in adolescents, perhaps due to an increased incidence of cerebral edema.100–102 In the experimental model of developmental concussion, the youngest rats became hypotensive after even mild injuries and tended to have longer apnea times than adult rats. After severe fluid percussion injury, mortality in these immature animals approached 100%.97
It is reasonable to hypothesize that diffuse mechanical injury can have lasting effects on the complex sequence of neurochemical and anatomical events occurring during normal development. Indeed, long-term follow-up studies demonstrate persistent neurocognitive deficits after pediatric TBI.103,104 However, it is also difficult to assess developmental deficits in children after mild brain injury, as signs of overt neurologic dysfunction may be lacking, and loss of developmental potential may only be demonstrable at a later time or under specific circumstances.
Environmental enrichment provides a useful experimental model with which to study developmental plasticity after brain injury. In an enriched environment, rats are reared communally in a large cage with multiple toys, tunnels, and objects that are changed regularly. When compared with animals reared in standard conditions, enriched rats have increases in cortical thickness, larger neurons, more glia, a greater number of synapses, and enhanced dendritic branching.105–107 The enriched rats are also superior in cognitive testing using the Morris water maze.108 However, when moderately concussed juvenile rats are reared in an enriched environment, they fail to develop the increased cortical thickness and enhanced cognitive performance seen in sham-injured enriched controls.109 These results demonstrate that developmental brain injury, even without early behavioral deficits or significant later morphologic damage, can lead to impaired plasticity. Further studies must be done to determine whether this loss of experience-dependent plasticity is permanent or whether it represents a window of impairment after which the capacity for neural reorganization recovers.
Overuse Injury
As demonstrated by the recent focus on concussion in football and hockey, both athletic trainers and athletes feel significant pressure to return athletes to practice and play as soon as possible after injury. Although returning to play may be delayed because of concerns about susceptibility to a second brain injury, returning to practice might seem like a reasonable means of maintaining physical conditioning while awaiting full recovery.
In animals, the importance of limb use in recovery of function after unilateral cortical lesions has been well demonstrated.110 In fact, recovery of function was associated with increased dendritic growth in the homotopic region of the uninjured cortex, dependent on use of the intact forelimb. However, restraint of the uninjured forelimb, and thus forced overuse of the injured limb, resulted in a failure of dendritic enhancement in the intact cortex, an increase in the lesion size in the injured cortex, and a longer-lasting behavioral deficit.111 There also appears to be a time window when the deleterious effects of forced overuse are mitigated to some degree. In the same model, when immobilization of the intact arm was delayed 1 week after the injury, the functional recovery was still delayed, but the increase in lesion size did not occur.112 The results of these studies suggest that, at least after focal brain injury, it is possible to overstimulate the injured brain and that this excessive activation can lead to longer-lasting deficits.
Repeated Concussion
How soon to return to play after a head injury and the consequences of repeated concussions are two of the most important health-related issues in sports today. We have earlier reviewed what is known about the neurometabolic cascade of events that occurs after experimental brain injury (Figure 1). Acute abnormalities include ionic fluxes, indiscriminate glutamate release, hyperglycolysis, lactate accumulation, and axonal injury. Later steps in this physiologic cascade involve increased intracellular calcium, mitochondrial dysfunction, impaired oxidative metabolism, decreased glycolysis, diminished CBF, axonal disconnection, neurotransmitter disturbances, and delayed cell death. It is during this postinjury period, when cellular metabolism is already stretched to its limits, that the cell is more vulnerable to further insults.13 Examining the time course of the post-TBI neurometabolic cascade may help us determine guidelines for vulnerability of the concussed brain to a second injury.
Several physiologic parameters indicate windows of potential vulnerability in the traumatized brain. First, consider the period of glucose metabolism-CBF uncoupling. This phenomenon is most dramatic during the hyperglycolytic phase, which, in the rat, begins at the time of injury and lasts for at least 30 minutes.22 At this time, cerebral metabolism is already at its limit, and any further demand in energy (due to increased ionic flux) or reduction in energy (due to impaired blood flow or reduced ATP synthesis) may tip the scale in favor of irreversible neuronal injury. Thus, injured cells may be capable of recovering after an initial injury, but a second concussion during this energy crisis can lead to cell death. After the initial hyperglycolytic period, cerebral glucose metabolism is reduced, as is CBF, apparently resolving the mismatch in energy supply and demand. However, during this period, CBF may be unable to respond to a stimulus-induced increase in cerebral glucose metabolism, reinstating the metabolic crisis. An increase in glycolysis in this period may be due to excessive external stimulation or a second injury (concussion, ischemia, or seizure).
A second potential period of vulnerability centers on intracellular Ca2+ accumulation. Increased Ca2+ levels may impair mitochondrial metabolism at the time when the cell can least tolerate a reduction in ATP production. Additional Ca2+ influx, again due to increased physiologic stimulation or a second injury, may go on to activate proteases that initiate the march to programmed cell death. In the rat model, this period of acute Ca2+ accumulation is somewhat severity dependent and lasts 2 to 4 days.50,52
Another period of risk may be associated with impaired neurotransmission. Alterations in NMDA receptor composition can persist for up to 1 week after injury in developing rats,84 and a second injury in this period can lead to further impairment of excitatory neurotransmission with a greater degree of cognitive dysfunction. Long-term potentiation, postulated as a mechanism for learning and memory, is impaired for up to 8 weeks after experimental brain injury91 and may be another means by which altered excitatory neurotransmission results in neurobehavioral deficits. Diminished attention and cognition are particularly important in an athletic setting, when subtle impairments will likely increase the risk of recurrent head injury.
Post-TBI changes in inhibitory neurotransmission seen in rats95,96 can leave neurons more susceptible to massive depolarization and EAA release after a recurrent concussion. Excessive excitation may then more easily lead to seizure activity, increased energy demand, and possibly further cell death.
As each of these physiologic parameters has its own time frame, and each head injury can be very different from the next, it is difficult to definitively state the true duration of vulnerability to a second injury. Preliminary studies using a double concussion model in rats revealed increased anatomic damage and prolonged hypometabolism when 2 concussions were separated by as much as 5 hours.114 Double concussion also appears to increase immunostaining for glial fibrillary acidic protein (a marker for gliosis and scarring) and lead to greater cell loss when compared with a single injury.115 Interestingly, in a recent report, multiple mild concussions preceding a more severe concussion by 3 to 5 days actually resulted in a better functional outcome than an isolated severe concussion.116 However, the anatomic injury appeared unchanged. This finding raises the possibility that after TBI, there is a period of increased danger to a second injury, followed by a period when the brain may actually be more capable of recovering from a repeated injury.
Of course, translating these experimental time frames into time frames relevant to human concussion can be tricky. Posttraumatic derangements in glucose metabolism resolve within 7 to 10 days in rats, whereas in humans, persistent depression of glucose uptake has been reported 2 to 4 weeks later.58 Evidence of secondary axotomy is seen as soon as 4 hours postinjury in animals, but evidence of ongoing axonal damage has been reported in human brain tissue even weeks after trauma.75 In general, the time frame for events in rats is much shorter than for similar periods in humans, and it would not be unreasonable to assume that these periods of postinjury physiologic change are longer lasting in humans. In addition, differences in injury type and severity certainly affect the duration of these neurometabolic changes and must also be considered when determining back-to-play status.
SUMMARY
Cerebral concussion is followed by a complex cascade of ionic, metabolic, and physiologic events. The earliest changes are an indiscriminate release of EAAs and a massive efflux of K+ triggering a brief period of hyperglycolysis. This is followed by more persistent Ca2+ influx, mitochondrial dysfunction with decreased oxidative metabolism, diminished cerebral glucose metabolism, reduced CBF, and axonal injury. Late events in the cascade include recovery of glucose metabolism and CBF, delayed cell death, chronic alterations in neurotransmission, and axonal disconnection. Clinical signs and symptoms of impaired coordination, attention, memory, and cognition are manifestations of underlying neuronal dysfunction, most likely due to some of the processes described above. It is difficult to match clinical signs with specific underlying physiologic derangements, and guidelines permiting return to play only after resolution of all motor and cognitive deficits are a minimal precaution. There is recent evidence that even relatively asymptomatic (GCS 13-15) patients may demonstrate depressed glucose metabolism on PET imaging following TBI, reinforcing the need for meticulous clinical assessment. Further study will reveal more details of the time course for this neurometabolic cascade. This will eventually permit improved clinical monitoring of posttraumatic pathophysiology in actual patients, including variables such as cerebral glucose metabolism, blood flow, neuronal activity, and even molecular changes.
Traumatic injury to the developing brain may lead to long-lasting changes in cognitive potential, perhaps even with little evidence of an initial deficit. Children and adolescents who sustain a concussive brain injury should be closely monitored over time for the later appearance of neurobehavioral abnormalities.
Repeated injury within a particular time frame can lead to a much larger anatomical or behavioral impairment than 2 isolated injuries. The second injury may be obvious, such as a repeated concussion, hypoxia, or seizure, or it may occur in the form of premature activation or overstimulation of the injured brain. An awareness and understanding of postconcussive pathophysiology will help in determining the best time course for return to practice and return to play.
ACKNOWLEDGMENTS
This work was supported by NS37365, NS30308, NS27544, and the Lind Lawrence Foundation.